Research Projects
The Persson Group studies the physics and chemistry of materials using atomistic computational methods and high-performance computing technology. Our main tools are density functional theory (DFT) and molecular dynamics (MD), but we also use a range of machine learning (ML) and natural language processing (NLP) methods to take advantage of large data sources. We collaborate heavily with experimental teams, ensuring that our materials make it out of the computer, onto the lab bench, and out into the world.The Materials Project
The Materials Project is a multi-institution, multi-national effort to compute the properties of all inorganic materials and provide the data and associated analysis algorithms for every materials researcher free of charge. The ultimate goal of the initiative is to drastically reduce the time needed to invent new materials by focusing costly and time-consuming experiments on compounds that show the most promise computationally.
The Materials Project has already performed more than 1 million calculations across 130,000 distinct inorganic compounds (requiring hundreds of millions of CPU hours), and has registered over 125,000 users from around the world. Furthermore, we are building tools to facilitate user-contributed data (both computational and experimental) to help researchers disseminate their data and further enrich the entire materials science community.
Leveraging the Materials Project data and capabilities, users have published papers on Li battery materials, solid state electrolytes, piezoelectric materials, and magnetic materials, amongst other applications. Several Python libraries developed by Materials Project are also released open source and are used by thousands of researchers around the world.
Within our group, we use the Materials Project to design novel materials for a diverse range of applications, from piezoelectrics to magnetocalorics and from transparent conductors to heterogeneous catalysts. As we design and study these materials, we are also constantly expanding the Materials Project database and analysis capabilities. Current work includes studying complex (non-ferromagnetic) magnetic orderings and developing workflows for surface studies.
Electrodes for Next-Generation Batteries
The electrodes (cathode and anode) of a battery determine its theoretical capacity, making the choice of electrode materials important for both current-generation Li-ion batteries and for next-generation battery technologies (including Li-Si and multivalent batteries). Even if a combination of anode and cathode have a high theoretical capacity, it is imperative that they be designed to prevent degradation, a process which can be chemical, electrochemical, and/or mechanical in nature. Our group designs and studies electrode materials for a range of battery chemistries, tackling both the challenges of capacity and stability.
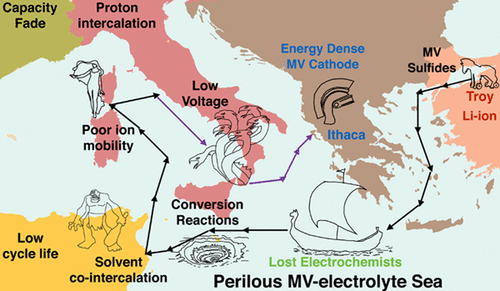
The Multivalent Cathode Screening project uses high-throughput DFT to search for new chemical compounds that can serve as electrodes by exchanging multivalent cations such as Mg2+, Ca2+, and Y3+. Such formulations can potentially offer large improvements in energy density over today’s Li+ based batteries, but suitable electrode compounds must be identified amongst tens of thousands of potential candidates.
Li-Si batteries, which use silicon instead of graphite as the battery anode, offer energy storage densities roughly 10 times as high as conventional Li-ion batteries. We use first-principles modeling, including DFT and ab initio molecular dynamics (AIMD), to study the thermodynamic and kinetic properties of various silicon-based anodes, including amorphous silicon, silica, and silicon-based alloys.
Surface coatings are widely used to protect battery cathodes and prevent degradation processes. We investigate amorphous coatings for Li-ion battery applications, studying their thermodynamics stability and Li+ diffusivities.
Dynamics of Battery Electrolytes
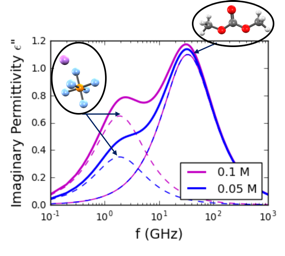
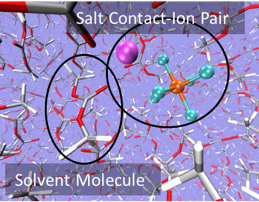
Liquid electrolytes must simultaneously provide favorable transport properties, allowing metal ions to conduct in response to applied voltage, and must be stable to prevent ongoing degradation. We study speciation, transport, electrochemistry, and reactivity in battery electrolytes for a range of different classes of batteries (Li-ion and Li-Si, as well as multivalent batteries).
Liquid electrolytes for Li-ion Batteries (LIB) can exhibit a variety of speciated salt complexes which are in equilibrium with each other. For example, free ions can be present alongside associated neutral pairs. We are interested in exploring speciation and solvation structure at an atomic and molecular level, as well as the consequences of speciation on electrochemical stability and transport properties. To date, we have explored a range of common Li-ion battery electrolytes, including LiPF6 in ethylene carbonate (EC) and in the "Gen2" electrolyte with ethyl methyl carbonate (EMC) and fluorethylene carbonate (FEC). We are also working to model novel classes of electrolyes, including superconcentrated electrolytes with high ion concentration and electrolytes designed for extreme low-temperature use.
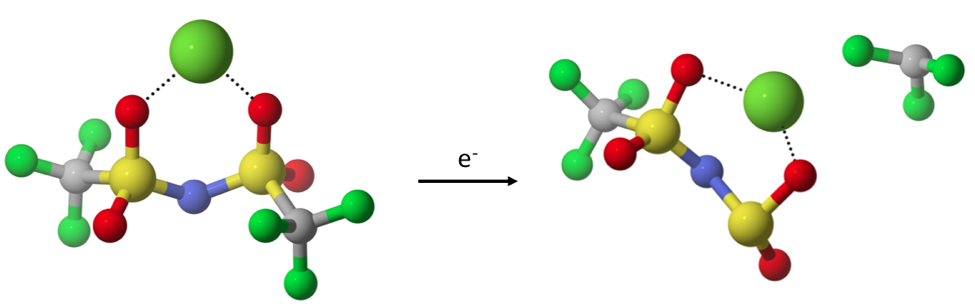
While multivalent cations show potential for energy storage devices with greater capacities than current lithium-based technologies, their practical implementation is inhibited by the lack of suitable electrolytes. Among the challenges posed by electrolytes for multivalent cation batteries is chemical stability during the multi-electron charge transfer processes involved in metallic plating and stripping. During plating, the solvated divalent ion has to undergo a transient, partially charged valence state; e.g. Mg+ or Ca+, as it approaches the metal surface. The process is fundamentally different as compared to monovalent systems (e.g. Li and Na), where no intermediate, reactive cation state exists, as a one-electron transfer is sufficient for plating. We seek to identify solvent molecules and ion-paired anions that are not susceptible to decomposition, potentially allowing for reversible plating and stripping. We are also interested in comparing the reactivity of electrolyte molecules coordinated with different multivalent cations.
Modeling Solid-Electrolyte Interphase Formation
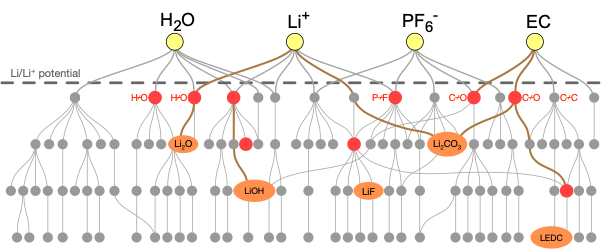
The solid-electrolyte interphase (SEI) is a nanoscale layer that forms on the anode of metal-ion batteries as a result of electrolyte decomposition. When properly formed, the SEI can conduct metal ions while preventing electron transmission, thereby preserving battery capacity. However, in certain cases, no SEI forms, causing runaway electrolyte degradation, or an SEI forms that prevents ion conductivity and therefore greatly reduces capacity. We seek to employ a range of techniques, including DFT, stochastic modeling, machine learning, and graph theory, to understand the reaction pathways involved in SEI formation.
We are simultaneously exploring SEI formation in Li-ion, Li-Si, and Mg-ion batteries. Using first-principles calculations, we construct massive chemical reaction networks, which we can then analyze using pathfinding algorithms and kinetic Monte Carlo approaches to obtain insights regarding various competing reaction mechanisms and SEI products. Our eventual goal is to use the insights gained from this reaction network approach to assist our experimental collaborators, for instance at the Joint Center for Energy Storage Research, in their efforts to control SEI formation through tailoring of the electrolyte and targeted reaction engineering. We also work closely with other theorists to develop complex SEI formation models at longer length scales.
Synthesizing New Materials
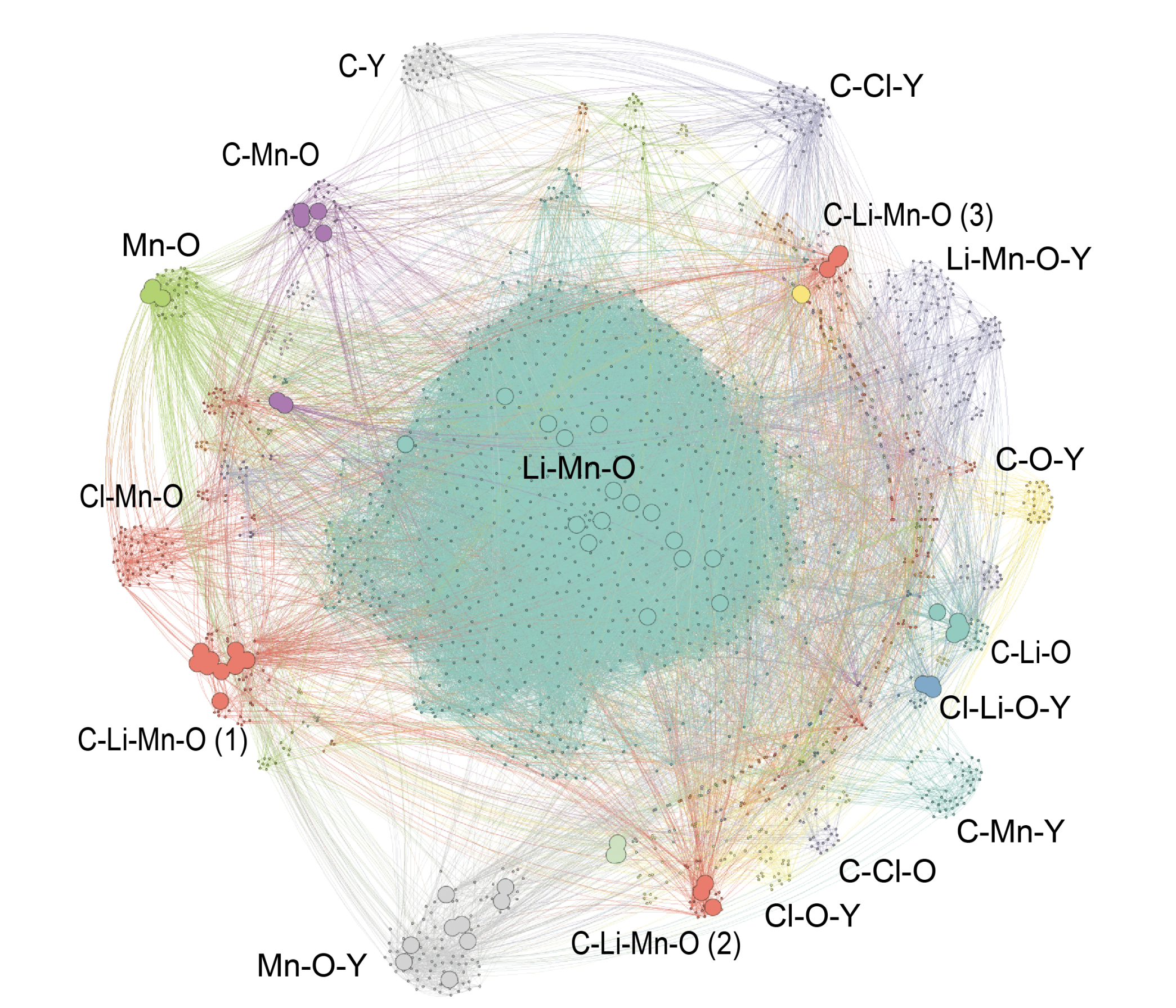
Computational analysis has identified countless materials that have yet to be synthesized experimentally. We seek to determine which of these theoretical materials can be synthesized, if they should be synthesized (based on their predicted properties), and how they can be synthesized.
One primary focus is dedicated to the enumeration of thermodynamic synthesis pathways in both liquid solution and solid state. In addition to the aqueous reaction pathways explored in the Solid-Electrolyte Interphase section above, solid state reaction networks are also studied using graph theoretical analysis.
In addition to reaction pathways, our group is applying Natural Language Processing (NLP) to identify the properties of materials through analysis of the existing materials science literature. This insight provides experimental efforts with a direction in the unlimited possibilities of material prototypes.
Exploiting Materials Symmetry
Through the mathematical framework of group theory, the study of symmetry has led to many fruitful discoveries and insights into physical laws that govern our world, from the link between conservation laws and symmetry operations to the entire field of crystallography. From the perspective of our group, describing a material in terms of its symmetries provides a tool for classifying materials, reducing computational cost, and screening materials for desired attributes.
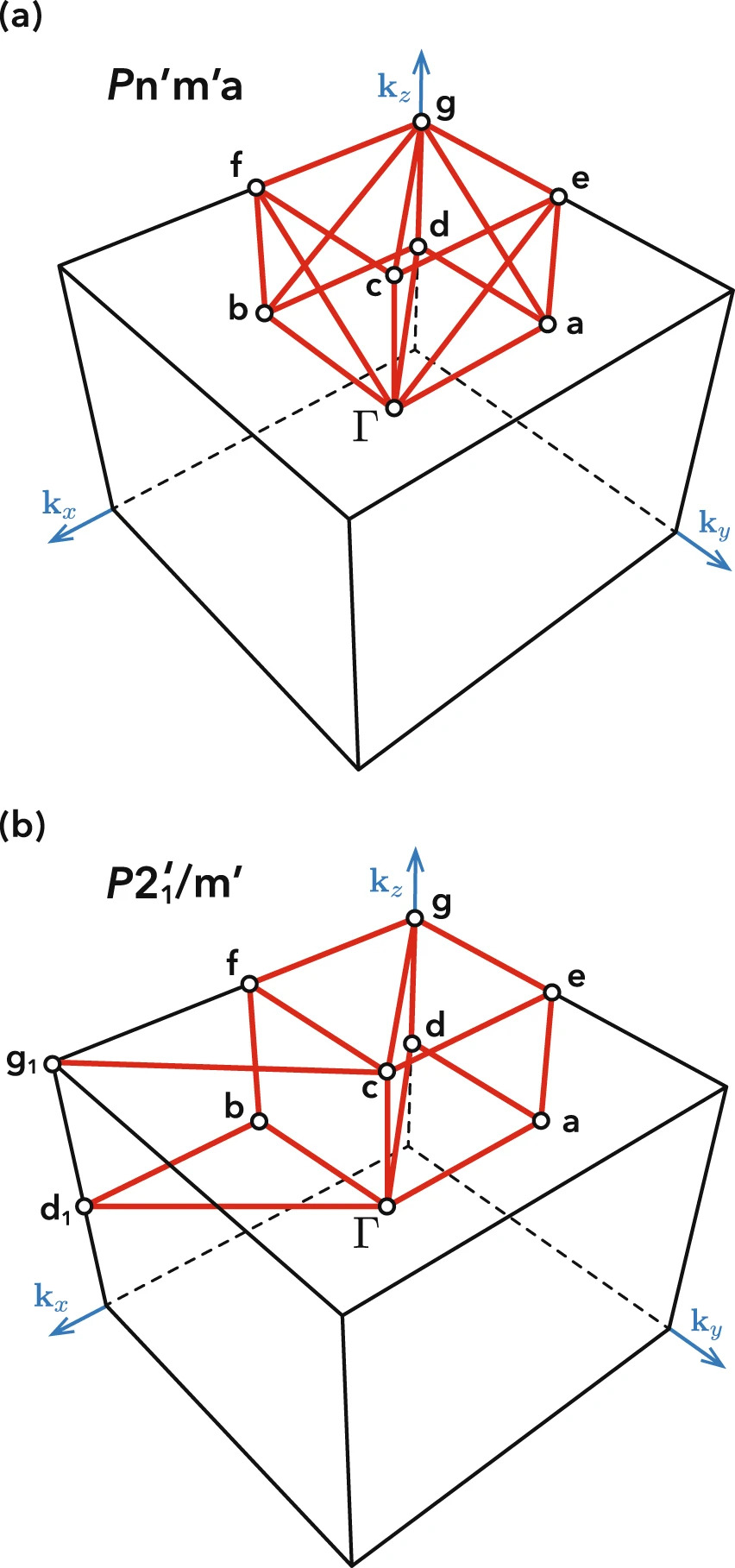
The symmetry of a material provides insight into its vibrational properties. Each crystal has a basis of phonon modes that obey the symmetry constraints. Understanding how these phonon modes relate to the underlying symmetry also lends itself to the study of structural phase transitions. One area of research includes the study of symmetrically permitted lattice distortions based on sublattice enumeration.
Reciprocal space symmetries of a material complement their real space counterparts and are important for the study of topological materials, which have some properties that are invariant under continuous deformation. A well-known example is topological insulators, which have symmetry-protected conductive states on their surfaces. Our group develops tools for classifying material band structures according to symmetry and topological order. Recently, we have developed a new method for determining the optimal choice of k-path to represent a material's band structure based on reciprocal representation of space groups (including magnetic space groups). In-depth symmetry analysis provides a useful description of electronic structure topology, as well as computationally efficient k-points sampling for high-throughput calculations.
In addition to topological properties, the magnetic symmetries of materials can be used to enumerate magnetic orderings permitted by crystallographic and time-reversal symmetries. We aim to develop computational workflows to calculate complex magnetic orderings and associated magnetic properties at scale, to enable discovery of new magnetic materials. One application of this research is discovery of new magnetocaloric materials, which are materials that undergo a temperature change under an applied magnetic field and can be used for room-temperature refrigeration.